
Last Updated on 12 mei 2024 by M.G. Sulman
Pulmonale hypertensie (PH) is een aandoening waarbij de bloeddruk in je longslagader te hoog is. Dit zorgt ervoor dat de rechterkant van je hart harder moet werken om bloed door de longen te pompen. Pulmonale hypertensie zelf is geen ziekte, doch eerder een symptoom van andere onderliggende aandoeningen. Denk hierbij aan problemen met de linker hartkamer, longweefsel of lever. Bepaalde onderliggende aandoeningen komen vaak voor, zoals problemen met de linker hartkamer of longweefsel, terwijl andere zeldzamer zijn, zoals aandoeningen van de longbloedvaten.
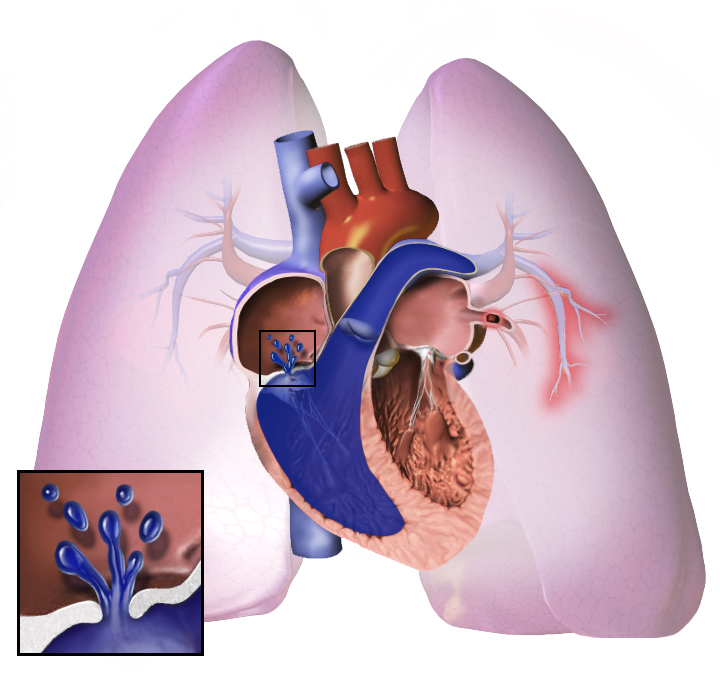
Wat is pulmonale hypertensie?
Pulmonale hypertensie is wanneer de bloeddruk in je longvaten te hoog is. Hierdoor heeft het rechterdeel van je hart moeite om bloed naar je longen te pompen. Dit kan leiden tot verwijding van het rechterdeel van je hart en verminderde pompkracht.
Vóórkomen
Het treft ongeveer 1% van de bevolking.
Wie krijgt het?
Pulmonale hypertensie kan mensen van alle leeftijden treffen, vooral zij met hart- of longproblemen. Het komt ook vaker voor bij mensen met andere gezondheidsproblemen, zoals:
- Ernstige problemen met de mitralisklep bij bijna iedereen.
- Bij ongeveer 65% van mensen met een probleem aan de aortaklep.
- Tot 30% van de mensen met sclerodermie.
- Bij ongeveer 20-40% van mensen met sikkelcelziekte.
- Ongeveer 1 op de 200 mensen met hiv.
PH kan soms ook pasgeborenen treffen, hetgeen persistente pulmonale hypertensie van de pasgeborene (PPHN) wordt genoemd. Baby’s met deze aandoening hebben mogelijk behandeling op de intensive care nodig.
Leeftijd
Pulmonale hypertensie kan op verschillende leeftijden worden gediagnosticeerd. Voor PAH wordt de diagnose meestal gesteld tussen de leeftijd van 41 en 60 jaar, met ongeveer 41% van de gevallen in die leeftijdsgroep en 36% tussen 61 en 80 jaar. Pulmonale arteriële hypertensie (PAH) is een zeldzame, progressieve vorm van PH. Bij PH als gevolg van CTEPH, een levensbedreigende vorm van pulmonale hypertensie, wordt de diagnose ook meestal na het 40e jaar gesteld, met ongeveer 30% van de gevallen tussen 41 en 60 jaar en 50% tussen 61 en 80 jaar. Er zijn geen specifieke cijfers bekend voor andere groepen.
Oorzaken van pulmonale hypertensie
Pulmonale hypertensie heeft vaak een onderliggende oorzaak, zoals hart-, long- of leverproblemen, systemische ziekten of bloedziekten. Soms kan het ook het gevolg zijn van een infectie. Er zijn meer dan 50 verschillende oorzaken bekend voor pulmonale hypertensie, waardoor het in 5 verschillende groepen is ingedeeld, elk met een andere specifieke oorzaak.
Onderzoek en diagnose
Hartkatheterisatie
Om de diagnose van pulmonale hypertensie vast te stellen, voert een arts uitgebreide onderzoeken uit. Allereerst wordt een hartkatheterisatie gebruikt om te bevestigen of er daadwerkelijk sprake is van pulmonale hypertensie. Tijdens een hartkatheterisatie wordt een dun slangetje (katheter) via de hals of lies in de bloedvaten ingebracht en naar de longbloedvaten geleid. Met een drukmeter kan de arts de druk in deze bloedvaten meten.
Waarden
Pulmonale hypertensie wordt meestal geclassificeerd op basis van de gemiddelde druk in de longslagader. Dit staat bekend als de pulmonale arteriële druk. De typische classificaties zijn:
- Normale druk in de longslagader: < 20 mmHg
- Milde pulmonale hypertensie: 20-24 mmHg
- Matige pulmonale hypertensie: 25-39 mmHg
- Ernstige pulmonale hypertensie: ≥ 40 mmHg
Andere onderzoeken
Daarnaast zijn andere onderzoeken nodig om de oorzaak van pulmonale hypertensie te achterhalen. Bijvoorbeeld bloedonderzoek, een elektrocardiogram (ECG), een röntgenfoto van de longen of een CT-scan van de longen, en een echocardiogram van het hart. Soms is een kort ziekenhuisverblijf noodzakelijk om deze uitgebreide onderzoeken snel en efficiënt uit te voeren.

Stadia
De stadia van pulmonale hypertensie worden vaak ingedeeld in vier functionele klassen door de Wereldgezondheidsorganisatie (WHO). Deze klassen zijn gebaseerd op de symptomen die je ervaart en hoe goed je dagelijkse activiteiten kunt uitvoeren. Naarmate de PH vordert, worden de symptomen duidelijker en kunnen ze je dagelijks leven beïnvloeden.
- Klasse 1: Je hebt geen symptomen en kunt je dagelijkse activiteiten normaal uitvoeren.
- Klasse 2: Je hebt geen symptomen als je rust, maar ervaart enig ongemak of kortademigheid bij routinematige activiteiten zoals huishoudelijke taken of traplopen.
- Klasse 3: Hoewel je je nog steeds goed voelt in rust, vind je het moeilijker om normale taken uit te voeren vanwege vermoeidheid of kortademigheid.
- Klasse 4: Je hebt symptomen, zelfs in rust, en deze verergeren wanneer je probeert normale activiteiten uit te voeren.
Behandeling van pulmonale hypertensie
De behandeling van pulmonale hypertensie is gericht op het verbeteren van de kwaliteit van leven, omdat genezing helaas niet mogelijk is. Het verloop van de ziekte is onvoorspelbaar en verschilt van persoon tot persoon. Dit zijn in vogelvlucht de behandelopties voor de twee hoofdtypen van PH:
- Pulmonale arteriële hypertensie (PAH):
- Calciumkanaalblokkers: Verlagen de bloeddruk in de longslagaders en het hele lichaam.
- Diuretica (plastabletten): Helpen overtollig vocht uit het lichaam te verwijderen.
- Zuurstoftherapie: Kan nodig zijn als het zuurstofgehalte in het bloed te laag is.
- Pulmonale vasodilatatoren: Ontspannen de longslagaders, verbeteren de bloedstroom en verminderen de druk op het hart.
- Chronische trombo-embolische pulmonale hypertensie (CTEPH):
- Anticoagulantia: Voorkomen de vorming van bloedstolsels.
- Ballon atriale septostomie (BAS): Een procedure om de druk in de longslagaders te verlichten.
- Ballonpulmonale angioplastiek (BPA): Een procedure om de longslagaders te verwijden.
- Oplosbare guanylaatcyclasestimulator (SGCS): Vertraagt de ziekteprogressie.
- Pulmonale endarteriëctomie (PEA): Een operatie om bloedstolsels uit de longen te verwijderen, de enige mogelijke remedie voor CTEPH.
Zuurstoftherapie, medicatie, dieetveranderingen, veranderingen in leefstijl en soms chirurgie kunnen ook worden aanbevolen. Dit is afhankelijk van de onderliggende aandoening(en).
Pulmonale vasodilatatoren zijn medicijnen die worden gebruikt bij de behandeling van PAH en CTEPH. Ze helpen de longslagaders te ontspannen, waardoor de bloeddruk daalt en de belasting van het hart verlicht wordt. Andere medicijnen kunnen worden gebruikt op basis van de specifieke onderliggende aandoeningen van de patiënt.
Prognose en levensverwachting
De vooruitzichten voor mensen met pulmonale hypertensie zijn afhankelijk van verschillende factoren, waraonder:
- Wat de oorzaak is van de PH.
- Hoe vroeg de ziekte wordt ontdekt en behandeld.
- Hoe ernstig de symptomen zijn.
- Eventuele andere medische aandoeningen die aanwezig zijn.
Elke persoon heeft een unieke situatie en prognose. Je levensverwachting kan verbeteren met vroege detectie en een passend behandelplan, maar het blijft een ernstige aandoening met een verhoogd risico op complicaties.
Reacties en ervaringen
Hieronder kun je reageren op dit artikel. Je kunt bijvoorbeeld je ervaringen delen over pulmonale hypertensie, of tips geven. Wij stellen reacties zeer op prijs. Reacties worden niet automatisch (direct) gepubliceerd. Dit gebeurt nadat ze door de redactie gelezen zijn. Dit om ‘spam’ of anderszins ongewenste c.q. ongepaste reacties eruit te filteren. Daar kunnen soms enige uren overheen gaan.

