
Last Updated on 1 maart 2024 by M.G. Sulman
Het Allan-Herndon-Dudley-syndroom (AHDS), ook bekend als MCT8-deficiëntie, is een zeldzame genetische aandoening die de ontwikkeling, mobiliteit en algehele gezondheid van kinderen ernstig beïnvloedt. Deze aandoening wordt veroorzaakt door een defect in het MCT8-schildklierhormoontransporteiwit. Dit leidt in het onvermogen van schildklierhormonen om de hersencellen binnen te dringen. Tegelijkertijd hoopt overtollig schildklierhormoon zich op in de rest van het lichaam, wat levensbedreigende complicaties tot gevolg kan hebben. Kinderen met AHDS worden geboren met een permanente en ernstige ontwikkelingsachterstand als gevolg van een tekort aan schildklierhormoon in de hersenen, terwijl ze tegelijkertijd hyperthyreoïdie ervaren in de rest van hun lichaam. Dit leidt tot verminderde spiertonus, ongecoördineerde bewegingen, rusteloze slaap, verhoogde hartslag, groeiachterstand en uiteindelijk ernstige complicaties zoals infecties en hart- en ademhalingsfalen.

Wat is het Allan-Herndon-Dudley-syndroom?
Het Allan-Herndon-Dudley-syndroom, ook bekend als MCT8-deficiëntie, is een erfelijke neuromusculaire aandoening die gepaard gaat met verstandelijke beperking en bewegingsproblemen door zwakke spierspanning en onderontwikkeling van spieren.
Naamgeving
Het Allan-Herndon-Dudley-syndroom is voor het eerst beschreven door Allan, Herndon en Dudley in 1944.1 Allan, William; Herndon, C. N.; Dudley, Florence C. (1944). “Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly”. American Journal of Mental Deficiency. 48: 325–34. Ze hebben in hun publicatie de kenmerken van het syndroom beschreven, inclusief de ernstige ontwikkelingsachterstand, lage spierspanning en bewegingsproblemen die gepaard gaan met deze genetische aandoening.
Vóórkomen
Het Allan-Herndon-Dudley-syndroom treft ongeveer 1 op de 70.000 mannen.
Oorzaak van MCT8-deficiëntie
MCT8-deficiëntie wordt veroorzaakt door een fout (mutatie) in het SLC16A2-gen, dat zich bevindt op het X-chromosoom. Het SLC16A2-gen codeert voor het MCT8-eiwit, een schildklierhormoontransporteur. Een fout in dit gen leidt tot het onvermogen van schildklierhormonen om effectief de hersencellen binnen te dringen. Dit leidt tot de kenmerkende symptomen van het syndroom, waaronder ernstige ontwikkelingsachterstand en neurologische problemen.
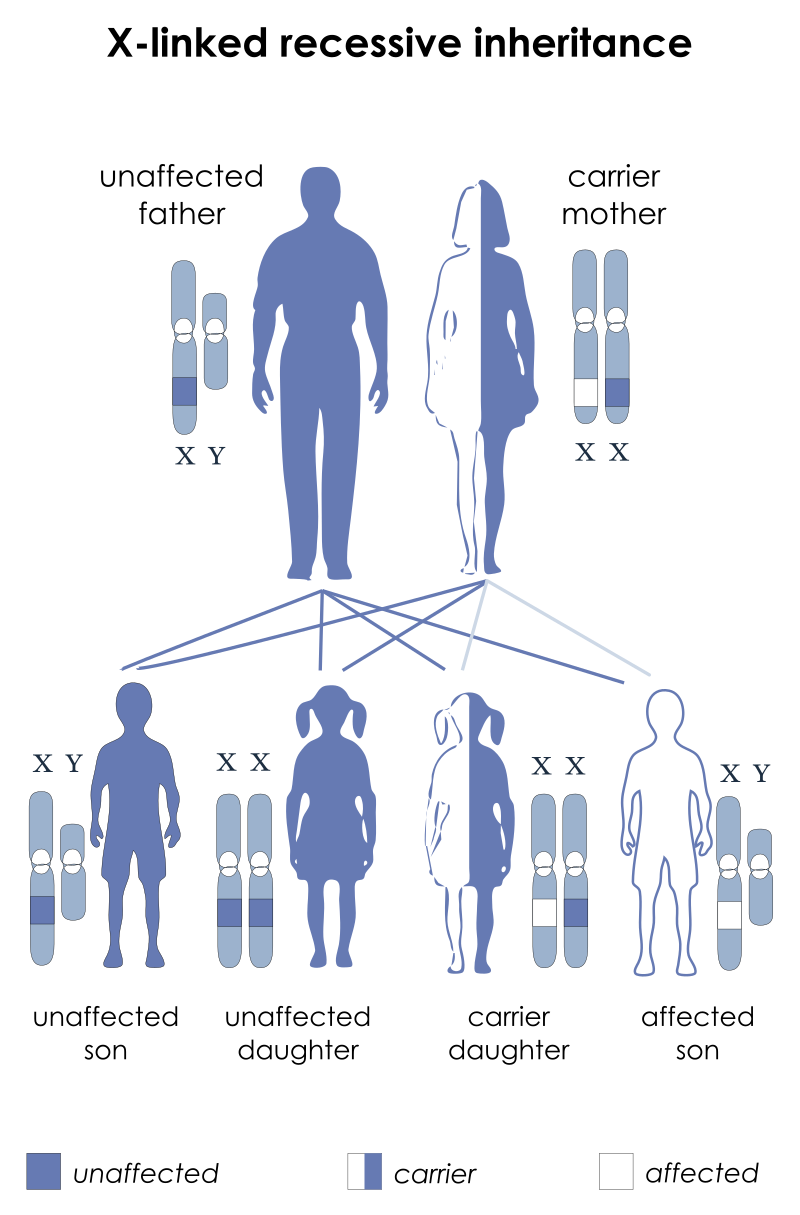
De aandoening is X-gebonden, wat betekent dat mannelijke baby’s een 50% kans hebben om de aandoening te erven als hun moeder draagster is van het afwijkende gen dat codeert voor het MCT8-schildklierhormoontransporteiwit.
Bij X-gebonden recessieve overerving kunnen de kenmerken als volgt worden samengevat:
- Moeder als draagster: De moeder van een patiënt heeft de aandoening zelf niet, maar is draagster van het abnormale gen dat codeert voor het MCT8-schildklierhormoontransporteiwit.
- Kans voor zonen: Zonen van draagsters hebben een 50% kans om het afwijkende gen te erven en daardoor de aandoening te ontwikkelen. Dit komt omdat ze één X-chromosoom van hun moeder erven, en als dat X-chromosoom het abnormale gen draagt, zullen ze de aandoening krijgen.
- Kans voor dochters: Dochters hebben ook een 50% kans om het afwijkende gen te erven. Dochters erven één X-chromosoom van hun vader en één X-chromosoom van hun moeder. Als het gezonde gen (niet het afwijkende gen dat verantwoordelijk is voor de aandoening) op het X-chromosoom van de vader aanwezig is, dan zullen dochters beschermd zijn tegen de aandoening. Echter, als het afwijkende gen zich op het X-chromosoom van de vader bevindt, dan zullen dochters draagster zijn van het afwijkende gen, maar ze zullen de aandoening zelf niet ontwikkelen.
Vrouwelijke dragers kunnen soms ook symptomen vertonen, variërend van mild tot ernstig. In sommige gevallen kan AHDS ook spontaan ontstaan tijdens de ontwikkeling van de foetus, waarbij het de eerste keer is dat de aandoening in een gezin voorkomt.
Symptomen MCT8-deficiëntie
MCT8-deficiëntie vertoont verschillende duidelijke tekenen en symptomen, waaronder:
- Uiterlijk: Patiënten kunnen een langer en smaller gezicht hebben in vergelijking met andere familieleden. Afwijkingen aan de wervelkolom (scoliose) en borstkas (pectus excavatum) kunnen ook aanwezig zijn.
- Ontwikkelingsachterstand en ernstige verstandelijke beperking: Het syndroom leidt tot een aanzienlijke ontwikkelingsachterstand, met onder mee het onvermogen om te spreken en te lopen.
- Hypotonie (lage spierkracht) en spasticiteit: Patiënten ervaren lage spierkracht (hypotonie) en stijfheid van de benen (spasticiteit).
- Onwillekeurige, ongeorganiseerde en abnormale spierbewegingen: Er zijn kortdurende, herhaalde uitbarstingen van verhoogde intensiteit die kunnen toenemen tijdens lichamelijk contact. In volwassenheid kunnen contracturen (dwangstand) en verminderd bewegingsbereik van de gewrichten optreden.
- Groeiachterstand/slechte gewichtstoename en ondervoeding: Patiënten vertonen problemen met gewichtstoename en voeding. Dit leidt tot een slechte groei.
- Verhoogde hartslag en abnormale hartslag: Tachycardie (verhoogde hartslag) en aritmie (abnormale hartslag) zijn ook kenmerkend voor MCT8-deficiëntie.
- Epilepsie: Een op de vier kinderen met het Allan-Herndon-Dudley syndroom ontwikkelt epilepsie, waarbij het optreden van verschillende soorten aanvallen sterk verbonden is met de leeftijd van het kind. Epileptische aanvallen manifesteren zich meestal in de vroege kinderjaren, zelden op latere leeftijd.
Onderzoek en diagnose
De diagnose van MCT8-deficiëntie wordt gesteld bij jongens die kenmerken vertonen zoals ontwikkelingsachterstand, verminderde spiertonus, verhoogde rusthartslag en slechte gewichtstoename. Meisjes die drager zijn van een fout in het SLC16A2-gen ervaren over het algemeen geen problemen, hoewel een klein aantal milde leerproblemen kan vertonen. De diagnostische stappen bestaan uit:
- Bloedtesten: Een bloedtest om het niveau van het schildklierhormoon T3 (liothyronine) te meten, evenals schildklierstimulerend hormoon (TSH) en T4 (thyroxine).
- Specifieke hormoonniveaus: Een verhoogd T3-niveau in combinatie met een normaal TSH-niveau en een laag normaal T4-niveau kan wijzen op AHDS.
- Genetische test: Teneinde de diagnose te bevestigen, zal er een bloedtest worden uitgevoerd om te zoeken naar genetische mutaties in het SLC16A2-gen, het gen dat verantwoordelijk is voor MCT8-deficiëntie.
- Specialistisch team: Een team van specialisten, waaronder een kinderneuroloog of endocrinoloog, samen met een geneticus en genetisch adviseur, zal betrokken zijn bij het diagnosticeren van AHDS en het informeren van de familie over het genetische aspect, met name de mogelijke overerving tussen generaties.
Behandeling van MCT8-deficiëntie
De behandeling van MCT8-deficiëntie kan bestaan uit:
Onderzoek naar alternatieve schildklierhormonen
Hedendaags onderzoek richt zich op schildklierhormoonvarianten, zoals TRIAC (tiratricol, Teatrois, Emcitate) en DITPA, die mogelijk de noodzaak van MCT8 omzeilen om de hersencellen te bereiken.
Vroege identificatie en behandeling
Het vroegtijdig identificeren van baby’s met MCT8-deficiëntie is van kardinaal belang om de schadelijke effecten op de hersenen zo veel als mogelijk te beperken. Dit vereist vroegtijdige interventie, idealiter in het eerste trimester van de zwangerschap. Doch de diagnose wordt helaas vaak pas na de geboorte gesteld.
Multidisciplinaire zorg
Er is geen genezing voor het syndroom. De behandeling richt zich op het optimaliseren van de ontwikkeling van het kind, het onder controle houden van epilepsie, en vroege detectie en behandeling van bijkomende problemen. De aanpak bestaat uit samenwerking met specialisten zoals kinderartsen, kinderfysiotherapeuten, kinderlogopedisten, diëtisten, revalidatieartsen, orthopedagogen, kinder- en jeugdpsychiaters, endocrinologen, en andere specialisten – afhankelijk van de individuele behoeften van het kind.
Behandeling van epilepsie
Medicijnen zoals diazepam, midazolam, lorazepam, clonazepam, natriumvalproaat, levetiracetam, clobazam, en zonisamide kunnen worden gebruikt om epilepsie te behandelen. Andere behandelingsopties omvatten een ketogeen dieet, nervus vagusstimulator, en methylprednisolon. Bij een ketogeen dieet eet je heel weinig koolhydraten.
Behandeling van spasticiteit
Baclofen, botuline toxine-injecties, dantrium, artane, benzodiazepines, en in sommige gevallen, een baclofenpomp, worden gebruikt om verhoogde spierspanning te verminderen.
Behandeling van dystonie
Medicijnen zoals Baclofen, Trihexyfenidyl, clonazepam, L-Dopa, gabapentine, tetrabenazine, botuline toxine-injecties, en in sommige gevallen, deep brain stimulation, kunnen dystonie verminderen.
Ondersteunende therapieën
Kinderfysiotherapie, kinderlogopedie, diëtetiek, kinderergotherapie, en orthopedagogiek worden gebruikt om de ontwikkeling te stimuleren en om te gaan met bijkomende problemen.
Educatieve ondersteuning
Speciaal onderwijs, begeleiding door een orthopedagoog, en mogelijkheid voor aanpassingen op school worden overwogen.
Medische monitoring
Regelmatige controles door tandartsen, oogartsen, en monitoring van bijkomende problemen zoals reflux, kwijlen, verstopping van de darmen, scoliose, en botontkalking worden uitgevoerd.
Preventieve maatregelen
Vitamine D en calciumsupplementen, antibioticaprofylaxe bij terugkerende infecties, en maatregelen om complicaties zoals reflux te voorkomen, kunnen door de arts worden overwogen.
Prognose
Patiënten met MCT8-deficiëntie vertonen variabele symptomen en hebben een divers verloop. Velen hebben een kortere levensverwachting als gevolg van ondervoeding en ernstige infecties. Ondanks uitgebreide zorg zullen de meeste patiënten nooit zelfstandig lopen of communiceren. Zij zullen afhankelijk blijven van verzorgers voor dagelijkse activiteiten.
Reacties en ervaringen
Hieronder kun je reageren op dit artikel. Je kunt bijvoorbeeld je ervaringen delen over MCT8-deficiëntie, of tips geven. Wij stellen reacties zeer op prijs. Reacties worden niet automatisch (direct) gepubliceerd. Dit gebeurt nadat ze door de redactie gelezen zijn. Dit om ‘spam’ of anderszins ongewenste c.q. ongepaste reacties eruit te filteren. Daar kunnen soms enige uren overheen gaan.

