
Last Updated on 5 februari 2024 by M.G. Sulman
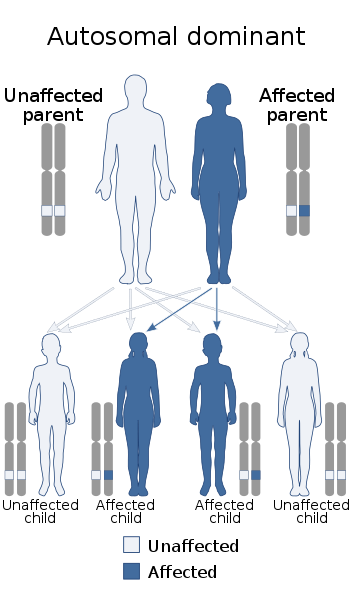
Myotone dystrofie type 1 (MD1) is een erfelijke spierziekte met verschillende mogelijke symptomen, die in de loop van de tijd vaak toenemen in ernst. Typerende kenmerken zijn het vertraagd ontspannen van aangespannen spieren, ook wel bekend als myotonie, en een langzaam toenemende spierzwakte, dystrofie genaamd. Naast spierproblemen kunnen ook andere organen klachten krijgen. Iemand met MD1 ervaart mogelijk vermoeidheid en een grotere behoefte aan slaap. Kinderen met MD1 hebben vaak leerproblemen en kunnen ook gedragsproblemen vertonen. MD1 is erfelijk en komt even vaak voor bij mannen als bij vrouwen. Als een van de ouders de ziekte heeft, is er bij elk kind opnieuw een risico van 50% om MD1 te erven. Een opvallend kenmerk van MD1 is dat binnen één familie de symptomen per generatie eerder beginnen en ernstiger worden, een fenomeen dat bekend staat als ‘anticipatie’.

Wat is myotone dystrofie type 1 (MD1)?
Myotone dystrofie type 1 is een zeldzame erfelijke ziekte die wordt gekenmerkt door het vertraagd ontspannen van aangespannen spieren, bekend als myotonie, en een geleidelijk toenemende zwakte van de spieren, dystrofie genaamd.
Naamgeving
Myotone dystrofie wordt vaak afgekort als ‘DM’, afkomstig van de Griekse naam dystrofia myotonica. De ziekte staat ook bekend als de ziekte van Steinert, genoemd naar de Duitse arts die de aandoening voor het eerst beschreef in 1909.
Betekenis
“Dystrofia myotonica” is afgeleid van het Grieks. De term bestaat uit twee delen:
- “Dystrofia” (δυστροφία) komt van het Griekse woord “dystrophia”, wat “slecht functionerend lichaamsweefsel” of “abnormale groei” betekent. In de context van spieraandoeningen verwijst het naar degeneratie of verslechtering van de spieren.
- “Myotonica” (μυοτονία) komt van het Griekse woord “myotonia”, wat verwijst naar het onvermogen van spieren om onmiddellijk te ontspannen na aanspanning. Het woord is samengesteld uit “mys” (spier) en “tonos” (spanning).
Samengevoegd verwijst “dystrofia myotonica” dus naar een aandoening die wordt gekenmerkt door progressieve spierdegeneratie (dystrofie) en het onvermogen van de spieren om snel te ontspannen na aanspanning (myotonie).
Vóórkomen
MD1 is een relatief zeldzame aandoenin. Naar schatting heeft één op de achtduizend mensen in West-Europa ermee te maken. In Nederland zijn er circa tweeduizend mensen met myotone dystrofie. 1Spierfonds. Wat is myotone dystrofie. https://www.spierfonds.nl/spierziekten/myotone-dystrofie (ingezien op 5-2-2024)
Vormen van myotone dystrofie
Er worden 4 vormen van myotone dystrofie type 1 onderscheiden.2Spierziekten Nederland. Vormen van myotone dystrofie (MD 1). https://www.spierziekten.nl/overzicht/myotone-dystrofie-type-1/vormen-van-myotone-dystrofie-md-1/ (ingezien op 5-2-2024)
1. Milde vorm
- Begint meestal na het vijftigste jaar.
- Klachten beperken zich vaak tot staar van de ooglens en mogelijk lichte spierzwakte.
2. Volwassen vorm (klassieke type)
- Vangt aan tussen twaalfde en vijftigste jaar.
- Langzaam toenemende spierzwakte, vooral in gezicht, kauw-, keel- en halsspieren, onderarmen en -benen.
- Beperkingen in lopen, traplopen, tillen, dragen van gewicht.
- Vermoeidheid, pijn, vertraagd ontspannen van spieren.
- Mogelijke stoornissen in diverse organen met toenemende leeftijd.
- Geen verschil in intelligentie vergeleken met gezonde mensen.
3. Kindervorm
- Klachten tussen het eerste en twaalfde levensjaar.
- Vertraagde verstandelijke ontwikkeling, spraakproblemen.
- Problemen met leren op basisschool, mogelijk speciaal onderwijs nodig.
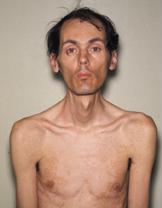
- Motorische ontwikkelingsachterstand, zwakke gelaatsspieren, uitdrukkingloos gezicht.
- Moeite in de omgang met andere kinderen, vermoeidheid, behoefte aan slaap.
- Buikpijn, verstopping of diarree, spraak- en slikproblemen.
4. Congenitale vorm
- Voornamelijk bij kinderen van moeders met MD1.
- Ernstige symptomen bij baby’s: slappe spieren, moeite met drinken en ademen.
- Verstandelijke en motorische ontwikkelingsachterstand.
- Vlakke gelaatsuitdrukking, ‘tentmondje’ (hierbij hangt de mond een beetje open en heeft de bovenlip een driehoekige vorm), en klompvoeten mogelijk.
- Buikklachten, oorontstekingen, spraakproblemen.
- Minder begaafd, leren wel spreken en verzorgen, maar moeite met leren lezen en schrijven.
Symptomen van myotone dystrofie type 1 (MD1) per vorm
Milde Vorm:
- Beginleeftijd: Na vijftigste jaar
- Vroege Symptomen: Staar, Myotonie
- Latere Symptomen: Lichte spierzwakte
Volwassen Vorm (Klassieke Type):
- Beginleeftijd: Tussen twaalf en vijftig jaar
- Vroege Symptomen: Myotonie, Spierzwakte, Maag- en darmklachten
- Latere Symptomen: Ernstige spierzwakte, Staar, Traagheid, Weinig initiatief, Slaperigheid, Orgaanstoornissen
Kindervorm:
- Beginleeftijd: Tussen één en twaalf jaar
- Vroege Symptomen: Leer- en gedragsproblemen, Spraakproblemen, Maag- en darmklachten
- Latere Symptomen: Myotonie, Spierzwakte, Gelijkaardig aan volwassen vorm
Congenitale (Aangeboren) Vorm:
- Beginleeftijd: Vóór de geboorte (foetus)
- Vroege Symptomen: Spierslapte, Ademhalings-, Slik- en Spraakproblemen, Klompvoetjes, Verstandelijke beperking
- Latere Symptomen: Myotonie, Spierzwakte, Gelijkaardig aan volwassen vorm
Oorzaak van MD1
Genetische oorzaak
Myotone dystrofie type 1 (MD1) wordt veroorzaakt door een fout in het DMPK-gen (dystrophia myotonica protein kinase) op chromosoom 19. Aan het einde van dit gen bevindt zich een stukje DNA bestaande uit groepen met een combinatie van bepaalde bouwstenen (C, T en G). Bij MD1 is er sprake van een teveel aan herhalingen van deze CTG-bouwstenen groep. De ernst van de aandoening hangt samen met het aantal herhalingen: des te meer herhalingen, des te ernstiger de symptomen.
Erfelijkheid en overdracht
MD1 is een autosomaal dominante aandoening. Dit betekent dat elk kind van een ouder met MD1 50% kans heeft om de ziekte ook te erven. Een opvallend kenmerk is dat binnen een familie de ziekteverschijnselen per generatie eerder beginnen en ernstiger zijn. Deze progressie komt voort uit de toename van de lengte van de CTG-herhalingen per generatie.
Onderzoek en diagnose
De diagnose van MD1 wordt doorgaans vastgesteld door een neuroloog op basis van kenmerkende symptomen zoals myotonie en spierzwakte. Myotonie is een neurologisch symptoom waarbij een langdurige aanspanning van de spier ontstaat bij percussie (oftewel ‘beklopping’). Het is echter ook mogelijk dat een andere medisch specialist de ziekte herkent.
De meest nauwkeurige methode voor het stellen van de diagnose is DNA-onderzoek, dat wordt uitgevoerd aan de hand van een buisje bloed. Dus bloedonderzoek.
In sommige gevallen kan de huisarts of specialist doorverwijzen naar een klinisch geneticus, een expert op het gebied van erfelijkheid. De klinisch geneticus speelt een waardevolle rol bij het stellen van de diagnose, het verstrekken van advies over het risico op de ziekte voor familieleden, en het beantwoorden van vragen met betrekking tot erfelijkheid en mogelijke kinderwens.
Behandeling van myotone dystrofie (MD1)
Anno 2024 bestaat er geen behandeling om MD1 te vertragen of te genezen. Onderzoek naar medicijnen is veelbelovend en er zijn allerlei met wetenschappelijke ontwikkelingen. De zorg richt zich op het vroegtijdig opsporen van complicaties, het optimaliseren van de conditie en kwaliteit van leven.
Genezing van MD1 is niet mogelijk, maar de gevolgen kunnen gelukkig draaglijker gemaakt worden en complicaties voorkomen. De conditie kan worden ondersteund en de leefomgeving aangepast. Een medicijn kan slaperigheid overdag helpen verminderen.
Gezien de wisselende en complexe symptomen, betrekt de begeleiding verschillende deskundigen. Reguliere controles bij een arts met expertise in MD1 zijn essentieel. Gespecialiseerde zorgverleners zijn te vinden in expertisecentra zoals die in Nijmegen en Maastricht.
Belangrijke Aandachtspunten bij MD1:
- Voor operaties: Informeer de chirurg en anesthesist vooraf, gezien het risico op complicaties tijdens en na algehele narcose. Draag het SOS-kaartje voor myotone dystrofie.
- Hartproblemen: Regelmatig een hartfilmpje (ECG) laten maken is aan te raden, vooral bij klachten als hartkloppingen en (bijna) flauwvallen.
- Ademhalingsproblemen: Falen van de ademhaling is een belangrijke oorzaak van klachten en overlijden. Ademhalingsondersteuning kan hierbij helpen.
- Zwangerschap: Specifieke medische begeleiding is noodzakelijk, aangezien het kind de ziekte ook kan erven.
Prognose en levensverwachting
De leeftijd waarop MD1 begint en de ernst van de symptomen variëren sterk. In milde gevallen manifesteert de ziekte zich mogelijk alleen met cataract en milde myotonie. Een ernstige presentatie kan echter levensbedreigende complicaties veroorzaken, zoals ademhalingsproblemen, slikproblemen of hartritmestoornissen. Ondanks het ontbreken van een remedie of behandeling die de progressie van MD1 kan vertragen, kan vroegtijdige interventie sommige complicaties verminderen of zelfs elimineren.
De vooruitzichten verschillen per vorm van MD1. Ongeveer één op de vijf kinderen met de aangeboren vorm overlijdt binnen enkele jaren, maar ongeveer de helft bereikt nog de veertig. Bij de kinder- en volwassen vorm is het gebruikelijk dat mensen tussen hun vijfenveertigste en vijfenzestigste levensjaar overlijden, vaak als gevolg van hart- of longproblemen. Mensen met de milde vorm hebben doorgaans een normale levensverwachting.
Reacties en ervaringen
Hieronder kun je reageren op dit artikel. Je kunt bijvoorbeeld je ervaringen delen over myotone dystrofie . Of tips geven. Wij stellen reacties zeer op prijs. Reacties worden niet automatisch (direct) gepubliceerd. Dit gebeurt nadat ze door de redactie gelezen zijn. Dit om ‘spam’ of anderszins ongewenste c.q. ongepaste reacties eruit te filteren. Daar kunnen soms enige uren overheen gaan.

